Die Diagnose Lungenfibrose gelingt am besten im interdisziplinären Team
 Die idiopathische pulmonale Fibrose ist durch eine progrediente Vernarbung des Lungengewebes unbekannter Ursache gekennzeichnet.
© Oranuch – stock.adobe.com
Die idiopathische pulmonale Fibrose ist durch eine progrediente Vernarbung des Lungengewebes unbekannter Ursache gekennzeichnet.
© Oranuch – stock.adobe.com
Die idiopathische pulmonale Fibrose (IPF) ist durch eine progrediente Vernarbung des Lungengewebes unbekannter Ursache gekennzeichnet. Sie galt lange als Paradebeispiel für eine rapide verlaufende und schlecht behandelbare interstitielle Lungenerkrankung (ILD). „Wenn ein Patient mit Exazerbation in der Klinik landet, beträgt die Dreimonatsmortalität 50 % – das ist eine wahnsinnig schlechte Prognose“, sagte Prof. Dr. Dirk Skowasch von der Klinik für Pneumologie am Herzzentrum Bonn.
Tatsächlich ist die IPF aber nicht mehr alleiniger Fokus: Der Begriff der progredienten pulmonalen Fibrose (PPF) umfasst inzwischen alle interstitiellen Lungenerkrankungen, die fortschreitend verlaufen. Es gibt bis zu 100 verschiedene Lungenfibrose-Subtypen, darunter zum Beispiel die Sarkoidose als die vielleicht häufigste Form, die aber sehr gut behandelbar ist.
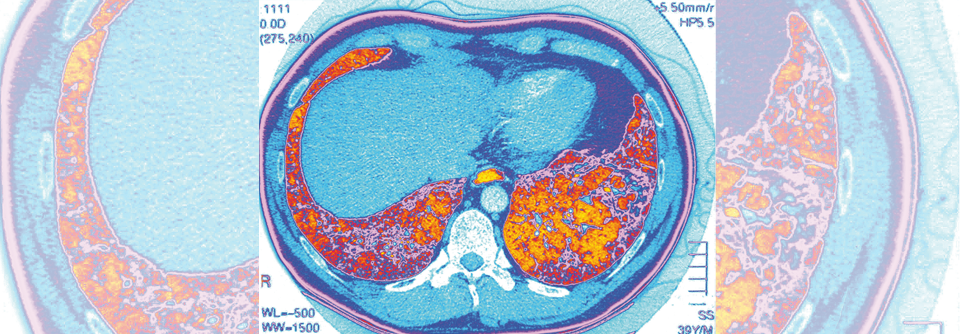
Die idiopathische pulmonale Fibrose ergibt sich diagnostisch durch Anamnese, Klinik, Serologie und hochauflösende Computertomografie (HRCT). Empfohlen wird eine native HRCT, also ohne Verwendung von Kontrastmittel, da dieses die typische Milchglastrübung als diagnostischen Hinweis maskieren kann. Im Zuge der Auskultation wies Prof. Skowasch auf die Besonderheit des Knisterrasselns hin. „Die Siderophonie hört sich an wie das Öffnen eines Klettverschlusses. Wenn man das einmal gehört hat, vergisst man das nicht mehr“, schilderte er. Auch das UIP*-Muster kann als Marker für eine idiopathische pulmonale Fibrose herangezogen werden. Ein weiterer Tipp des Pneumologen: „Wenn es sich nicht um einen älteren Mann handelt, der früher viel geraucht hat, spricht das gegen eine IPF.“
Interstitielle Lungenerkrankungen mit unklarer Genese erfordern in vielen Fällen weitere Diagnostik. Meist kommen dann die bronchoalveoläre Lavage oder bioptische Verfahren wie die transbronchiale Kryobiopsie zum Einsatz. Hierdurch können chirurgische Eingriffe vermieden werden. „Bei uns entscheidet das ILD-Board, ob eine Biopsie notwendig ist“, so Skowasch. Die Studienlage untermauert, dass hierdurch gerade bei unklassifizierten interstitiellen Lungenerkrankungen die Diagnosesicherheit entscheidend verbessert werden kann. Immer wieder hebt der Experte die Bedeutung des multidisziplinären ILD-Boards bei der Diagnose hervor. Idealerweise ist dieses besetzt mit Kolleginnen und Kollegen aus Pneumologie, Radiologie, Rheumatologie und Pathologie.
Überlebenszeit lässt sich durch einen schnellen Therapiestart von zweieinhalb auf bis zu acht Jahre verlängern
Unterstützung bei der Diagnostik des Krankheitsbilds bietet auch die Digitalisierung. KI-gestützte Bildanalyseprogramme zur Fibrosequantifizierung halten laut Skowasch bereits Einzug in den klinischen Alltag – inklusive Vorhersagen zur Mortalität. „Irgendwann werden wir den Radiologen gar nicht mehr brauchen“, prognostizierte Prof. Skowasch.
Bei bestätigter IPF ist der Start einer antifibrotischen Therapie mit dem Tyrosinkinaseinhibitor Nintedanib oder dem Immunsuppressivum Pirfenidon leitliniengerecht unmittelbar nach Diagnosestellung geboten. „Warten, ob der Patient einen Progress oder eine Exazerbation entwickelt – das sollte man heute nicht mehr“, betonte Skowasch. Studiendaten belegen: Die Überlebenszeit lässt sich durch einen schnellen Therapiestart von zweieinhalb auf bis zu acht Jahre verlängern. Zudem stehen auch neue Substanzen in den Startlöchern. Eine davon ist Nerandomilast, ein selektiver Inhibitor der Phosphodiesterase 4B. In Studien erzielte der Wirkstoff eine deutliche Verbesserung der Vitalkapazität bei Personen mit idiopathischer Lungenfibrose.
Bei der Therapie der progredienten pulmonalen Fibrose gilt: Erst, wenn andere für die jeweilige Diagnose angemessene Behandlungen keine ausreichende Wirksamkeit gezeigt haben, wird mit einer antifibrotischen Therapie begonnen. So sollte bei zugrundeliegenden rheumatischen Erkrankungen zunächst eine antiinflammatorische Therapie oder bei einer exogen allergischen Alveolitis eine Expositionskarenz versucht werden.
Im Gegensatz zur IPF ist bei der Behandlung der PPF nur Nintedanib als antifibrotische Option zugelassen. Die Therapieeinleitung soll im ILD-Board festgelegt werden. Ist Nintedanib nicht wirksam, kommt Pirfenidon zum Einsatz – allerdings off-label.
„Lungenfibrose ist nicht gleich Lungenfibrose“, resümierte der Pneumologe. Die Diagnostik sei komplex, die Erkrankungen heterogen – und Therapieentscheidungen müssten individuell getroffen werden. Nur ein strukturierter, interdisziplinärer Ansatz garantiert eine bestmögliche Versorgung der Patientinnen und Patienten mit interstitiellen Lungenerkrankungen.
* Usual Interstitial Pneumonia
Quelle: Kongressbericht – 131. Kongress der Deutschen Gesellschaft für Innere Medizin
Falls Sie diesen Medizin Cartoon gerne für Ihr nicht-kommerzielles Projekt oder Ihre Arzt-Homepage nutzen möchten, ist dies möglich: Bitte nennen Sie hierzu jeweils als Copyright den Namen des jeweiligen Cartoonisten, sowie die „MedTriX GmbH“ als Quelle und verlinken Sie zu unserer Seite https://www.medical-tribune.de oder direkt zum Cartoon auf dieser Seite. Bei weiteren Fragen, melden Sie sich gerne bei uns (Kontakt).

 Die idiopathische pulmonale Fibrose ist durch eine progrediente Vernarbung des Lungengewebes unbekannter Ursache gekennzeichnet.
© Oranuch – stock.adobe.com
Die idiopathische pulmonale Fibrose ist durch eine progrediente Vernarbung des Lungengewebes unbekannter Ursache gekennzeichnet.
© Oranuch – stock.adobe.com