primäre Vaskulitiden
Bei primären Vaskulitiden handelt es sich um immunreaktiv ausgelöste Gefäßentzündungen mit Schädigung der von den betroffenen Gefäßen versorgten Organe. Das Spektrum der klinischen Symptome hängt dabei von Ausmaß und Lokalisation der betroffenen Organe ab.
Die Vaskulitiden können nach der Größe der (überwiegend) betroffenen Gefäße eingeteilt werden:
Vaskulitis der großen Gefäße
Riesenzellarteriitis (RZA) → siehe dort
Takayasu-Arteriitis (TA)
- granulomatöse Entzündung der Aorta und ihrer Hauptäste
- Manifestation meist vor dem 40. Lebensjahr
- in Europa und Nordamerika selten (Inzidenz < 1/100.000/Jahr), gehäuft in China, Indien, Japan, Korea, Thailand, Afrika, Südamerika
- Frauen häufiger betroffen als Männer (w : m = 9:1)
- Prognose mit Therapie günstig, unbehandelt drohen KHK, Herzinfarkt, Schlaganfall, PAVK und weitere Gefäßkomplikationen
Vaskulitis der mittelgroßen Gefäße
Befallen sind vor allem die Hauptviszeralarterien und ihre Äste – es können aber im Prinzip Gefäße aller Größen betroffen sein.
Klassische Polyarteriitis nodosa (cPAN)
- nekrotisierende Arteriitis der mittleren und kleinen Arterien (ohne Glomerulonephritis und Befall von Arteriolen, Kapillaren, Venolen)
- keine Association mit ANCA (antineutrophiler cytoplasmatischer Antikörper)
- Inzidenz etwa 5/100.000/Jahr, m : w = 3:1
- Ätiologie unbekannt, gehäuft bei Hepatitis-B-Infektion
- Prognose ohne Therapie schlecht, mit Behandlung 5-Jahres-Überlebensrate bei etwa 90 %
Kawasaki-Syndrom
- Vaskulitis vor allem der mittelgroßen und kleinen Gefäße – aber auch Aorta und große Gefäße können betroffen sein
- häufig Beteiligung der Koronarien
- Auftreten typischerweise im Kindesalter (80 % der Patienten < 5 Jahre, häufigste Vaskulitis im Kleinkindesalter)
- Ätiologie unbekannt
Vaskulitis kleiner Gefäße
ANCA-assoziierte Vaskulitiden der kleinen Gefäße
Das sind idiopathische multisystemische Erkrankungen bei denen es durch die Bildung von ANCA zur destruktiven Inflammation überwiegend an kleinen Gefäßen kommt. Dazu gehören:
Wegenersche Granulomatose (Syn. Granulomatose mit Polyangiitis)
- nekrotisierende Vaskulitis von überwiegend kleinen und mittelgroßen Gefäßen mit verkäsenden Granulomen im Bereich des Respirationstraktes (Nase, Nebenhöhlen, Mittelohr, Oropharynx, Lunge)
- in 70 % Nierenbeteiligung (nekrotisierende Glomerulonephritis)
Mikroskopische Polyangiitis (MPA)
- Nekrotisierende Vaskulitis kleiner „mikroskopischer“, aber auch größerer Gefäße
- keine Granulome
- Inzidenz etwa 1/100.000/Jahr, m : w = 3:1
- 70 % Nierenbeteiligung (bestimmt die Prognose)
Churg-Strauss-Syndrom (eosinophile Granulomatose mit Polyangiitis –EPGA)
- granulomatöse nekrotisierende Vaskulitis vorwiegend der kleinen und mittelgroßen Gefäße des Respirationstraktes mit eosinophiler Infiltration des extravaskulären Gewebes
- Assoziation mit allergischem Asthma (bis zu 90 % der Patienten) und Eosinophilie
- sehr selten (Inzidenz 0,1/100.000/Jahr)
- Manifestation im mittleren Lebensalter
Nicht-ANCA-assoziierte Vaskulitiden der kleinen Gefäße
Immunkomplex-Vaskulitis
- Vaskulitis mit Ablagerung von Immunglobulinen und/oder Komplement in den Gefäßwänden
Anti-GBN (Glomeruläre Basalmembran)-Krankheit (Goodpasture-Syndrom)
IgA-Vaskulitis (Purpura Schönlein-Henoch – PSH)
- Ablagerung von IgA1-Immunkomplexen vor allem in den kleinen Gefäßen der Haut und des Darms
- häufig begleitet von Arthritis, manchmal Glomerulonephritis (IgA-Nephritis)
- meist Kinder im Vorschulalter (definitionsgemäß vor dem 21. Lebensjahr)
- allergische Vaskulitis der kleinen Blutgefäße und Kapillaren im zeitlichen Zusammenhang mit vorausgegangenem Atemwegsinfekt (50 % Influenza)
Kryoglobulinämische Vaskulitis
- Vaskulitis durch in der Kälte präzipitierende Immunglobulinkomplexe
Hypokomplementämische urtikarielle Vaskulitis
- seltene Kleingefäß-Vaskulitis mit Urtikaria und Komplementerniedrigung
Vaskulitiden variabler Gefäßgröße
Cogan-Syndrom
- entzündliche Affektionen von Augen, Innenohr, Vestibularissystem
- Vaskulitis von Gefäßen aller Größen (inklusive Aorta → Aortenaneurysma)
- entzündliche Beteiligung von Aorten- und Mitralklappen
Morbus Behcet
- Multisystemerkrankung mit leukozytoklastischer Vaskulitis
- einzige Vaskulitis mit dem Befall von Arterien und Venen
- in 70 % mit HLA-B51 assoziiert
- Erstmanifestation 20.–40. Lebensjahr, m : w = 3:1
- gehäuft in der Türkei, Prävalenz bei in Deutschland lebenden Türken 20/100.000

Die Symptome der Vaskulitiden hängen von Ausmaß und Lokalisation der Gefäßbeteiligung und der jeweiligen Art der Vaskulitis ab. Das klinische Bild kann sehr variabel sein, was die Diagnose erschwert.
Riesenzellarteriitis → siehe dort
Takayasu-Arteriitis
- schleichender Beginn über Jahre mit Fieber, Müdigkeit, Gewichtsverlust, Arthralgien, evtl. Kopfschmerzen
- im okklusivem Stadium (Pulseless-Phase) Pannikulitis, Erythema nodosum, Raynaud-Syndrom, Claudicatio intermittens (meist Arme)
- bei zerebraler Beteiligung Sehstörungen, Gesichtsfeldausfälle, Konzentrationsstörungen, Synkopen, Schlaganfall
- Symptome wie bei KHK und den jeweiligen Organinfarkten
Klassische Polyarteriitis nodosa (cPAN)
- Allgemeinsymptome (Fieber, Gewichtsverlust, Nachtschweiß)
- Muskel- und Gelenkschmerzen (60 %)
- gastrointestinale Beteiligung (50 %) → kolikartige Bauchschmerzen, evtl. Darminfarkte
- Hodenschmerzen
- Beteiligung der Koronarien (80 %) → Angina pectoris, Herzinfarkt in jungem Alter
- Beteiligung der Hirngefäße → Schlaganfall in jungem Alter
- Polyneuropathie (60 %), Mononeuritis multiplex, Epilepsie, Psychose
Kawasaki-Syndrom
- septische Temperaturen (> 5 Tage), die nicht auf Antibiotika ansprechen
- Konjunktivitis (meist beidseitig) mit verstärkter Gefäßinjektion
- Stomatitis (Rachenrötung und Erdbeerzunge ähnlich wie bei Scharlach)
- Palmar- und Plantaerythem → nach 2–3 Wochen Schuppung
- polymorphes rumpfbetontes Exanthem
- zervikale Lymphadenopathie (bei 50 %)
- Aneurysmen der Herzkranzgefäße, Koronaritis → Herzinfarkte
Wegenersche Granulomatose (Syn. Granulomatose mit Polyangiitis)
Lokalisiertes Stadium:
- chronische Rhinitis/Sinusitis (> 90 %), evtl. mit blutig-borkigem Schnupfen, Sattelnase, Septumperforation
- chronische Otitis (evtl. auch Mastoiditis)
- Ulzerationen im Oropharynx
- Lungenrundherde (60 %), evtl. mit Einschmelzungen (Pseudokavernen)
Generalisiertes Stadium:
- renale Beteiligung oder andere organbedrohende Manifestationen
- evtl. alveoläre Hämorrhagien mit Hämoptoe
- (rapid progressive) Glomerulonephritis → Gefahr des Nierenversagens
- evtl. Episkleritis, Arthralgien, Myalgien, ZNS-Symptome, periphere Neuropathien
- Fieber Gewichtsverlust, Nachtschweiß
Mikroskopische Polyangiitis (MPA)
- häufig Nierenbeteiligung (70 %) →Glomerulonephritis, renaler Hochdruck, Niereninsuffizienz
- pulmonale Vaskulitis (25 %) → evtl. alveoläre Hämorhagie mit Blut im Sputum
- Hautveränderungen (40 %) → subkutane Knötchen, palpable Purpura, evtl. Nekrosen
- weitere Symptome: Polyneuritis, Sinusitis, Episkleritis u.a.
Churg-Strauss-Syndrom (eosinophile Granulomatose mit Polyangiitis –EPGA)
- allergisches Asthma (bis zu 90 %), evtl. auch allergische Rhinitis, polypöse Sinusitis
- flüchtige Lungeninfiltrate
- evtl. Fieber
- kardiale Beteiligung (30 %) → Myokarditis, Koronaritis
- Mono/Polyneuropathie (75 %)
- ZNS-Vaskulitis (15 %)
- gehäuft Thromboembolien
- Nierenbeteiligung (20 %)
Immunkomplex-Vaskulitis
- vor allem Glomerulonephritis
Anti-GBN (Glomeruläre Basalmembran)-Krankheit (Goodpasture-Syndrom)
- rasch progrediente Niereninsuffizienz
- Lungenblutung
IgA-Vaskulitis (Purpura Schönlein-Henoch – PSH)
- Fieber und schweres Krankheitsgefühl
- Hautmanifestation (100 %) → Petechien und Exantheme („tastbare Purpura“,besonders Streckseiten der Beine und Gesäß)
- Gelenke (65 %) → schmerzhafte Schwellungen Sprunggelenke und andere Gelenke
- Gastrointestinaltrakt (50 %) → kolikartige Bauchschmerzen, Erbrechen, evtl. gastrointestinale Blutungen
- Nieren (klinisch 30 %, bioptisch 80 %) → Mikro- und Makrohämaturie
- ZNS → Kopfschmerzen, Verhaltensstörungen, pathologisches EEG
Kryoglobulinämische Vaskulitis
- akral betonte palpable Purpura
- Arthralgien
- Glomerulonephritis → Hämaturie, Proteinurie-Neuropathie
Hypokomplementämische urtikarielle Vaskulitis
- chronische Urtikaria mit Beteiligung anderer Organe
Cogan-Syndrom
- Augenmanifestation → rotes Auge, Photophobie, Verschwommensehen
- Menière-artige Anfälle mit Schwindel, Ataxie, Übelkeit, Erbrechen, Hörverlust
Morbus Behcet
- Haut/Schleimhäute → orale Aphthen (95 %), genitale Aphthen (70 %), Polyfollikulitis, Papulopusteln, Erythema nodosum
- Augenbeteiligung (80 %) → anteriore und/oder posteriore Uveitis, evtl. Optikusneuritis
- Arthritis (bis 70 %)
- Magen/Darm (bis 30 %) → Granulome, Ulcera, Perforationen
- ZNS (bis 30 %) → ZNS-Vaskulitis, Hirnstammsymptomatik, Sinusvenenthrombose
- Thromboembolien (korrelieren mit Krankheitsaktivität)

Der Untersuchungsbefund hängt von den jeweiligen Gefäßmanifestationen und den für die einzelnen Krankheitsformen typischen Symptomen ab.
- Identifikation von systemischen und lokalen Entzündungszeichen und Merkmalen von Ischämien und deren Regulationsmechanismen (insbesondere Strömungsgeräusche)
- pathologische Befunde bei der detaillierten Blutdruckmessung
Typische Laborbefunde
Riesenzellarteriitis:
BSG↑↑, CRP↑
Takayasu-Arteriitis:
BSG ↑↑, Anämie, Leukozytose
Klassische Polyarteriitis nodosa (cPAN):
BSG/CRP↑, Leuko-/Granulozytose, evtl. Thrombozytose, evtl. Komplement↓, evtl. positive Hepatitis-B-Serologie (25 %), ANCA-negativ
Kawasaki-Syndrom:
BSG/CRP↑, α2-Globuline↑, Leukozyten↑, Thrombozyten↑, Endothelzell-Antikörper (AECA) positiv
Wegenersche Granulomatose (Syn. Granulomatose mit Polyangiitis):
Oft BSG↑, Erythrozyturie, Serumkreatinin↑, evtl. Leukozytose, Thrombozytose, Anämie, cANCA (cytoplasmatic antibodies against neutrophil cytoplasmic antibody) positiv (meist gegen Proteinase 3 – PR3-ANCA)
Mikroskopische Polyangiitis (MPA):
pANCA (perinuclear antineutrophil cytoplasmic antibody) positiv (meist gegen Myeloperoxidase – MPO-ANCA)
IgA-Vaskulitis (Purpura Schönlein-Henoch – PSH):
Nachweis zirkulierender Immunkomplexe, Komplement anfangs oft erhöht, IgA↑
Hypokomplementämische urtikarielle Vaskulitis:
Komplement im Serum↓, ANA (antinukleärer Antikörper) positiv
Morbus Behcet:
HLA-B51-Nachweis (70 %), Ausschluss Virusinfektionen (Hepatitis B, C, HIV, HSV)
Weitere Untersuchungen hängen von den jeweiligen Manifestationen ab. Dazu können z.B. gehören:
- Angiographie (ggf. MR-Angiographie mit Gefäßwanddarstellung)
- Doppleruntersuchungen
- Koronarangiographie
- Ultraschall der abdominellen Organe (insbesondere Niere)
- Gewebebiopsien mit histologischer Klassifikation
- augenärztliche Untersuchung
- MRT, CT
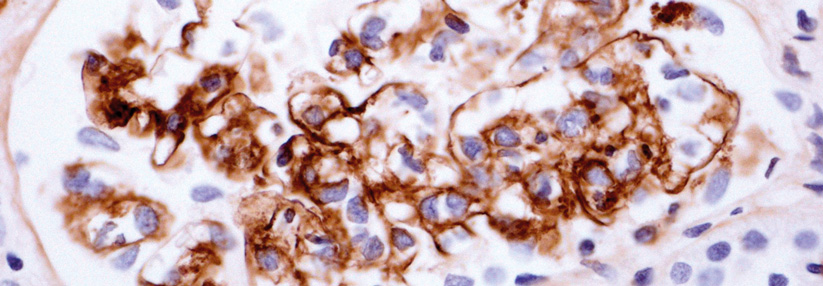
Aufgrund des bunten Bildes kommen zahlreiche Differenzialdiagnosen in Betracht. Dazu gehören:
- andere (nicht-vaskulitische) entzündliche Erkrankungen (z.B. MS bei ZNS-Beteiligung, rheumatoide Arthritis)
- andere Gefäßerkrankungen (z.B. Arteriosklerose)
- jeweils andere Formen der Vaskulitis
- reversibles Vasokonstriktionssyndrom (als DD zur zerebralen Vaskulitis)

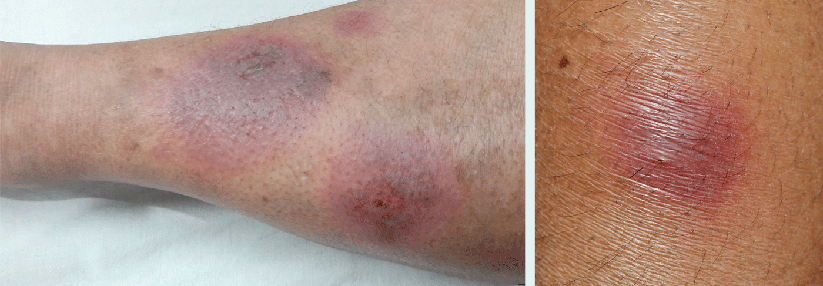
Grundlage der Therapie der primären Vaskulitiden ist die effektive Immunsuppression. In der Regel erfolgt eine Induktionstherapie, gefolgt von einer mindestens 18-monatigen Erhaltungstherapie.
Riesenzellarteriitis: → siehe dort
Takayasu-Arteriitis
- Glukokortikoide, Methotrexat (MTX)
- als Reservemittel: Cyclophosphamid, TNFα-Inhibitoren
- ASS
- ggf. Stenosebeseitigung (immer unter Immunsuppression)
Klassische Polyarteriitis nodosa (cPAN)
- Hepatitis-B-assoziiert: antivirale Therapie + Steroidtherapie ± Plasmaaustausch
- ohne Hepatitis B: MTX, bei progredientem Verlauf Cyclophosphamid + Steroide
Kawasaki-Syndrom
- hochdosierte Immunglobuline i.v. + ASS (ASS hier auch bei Kindern)
- ggf. intensivierte Thrombozytenaggregationshemmung
Wegenersche Granulomatose (Syn. Granulomatose mit Polyangiitis)
Lokalisiertes Stadium:
- Induktionstherapie MTX und Steroide
- Erhaltungstherapie: niedrig dosierte Steroide und Azathioprin (oder Leflunomid, MTX)
Generalisiertes Stadium (mit renaler Beteiligung):
- Induktionstherapie: hochdosiertes Prednisolon (bei schweren Verläufen auch als Bolustherapie plus Cyclophosphamid)
- Rituximab als Alternative
- Erhaltungstherapie (mindestens 18 Monate): Azathioprin oder MTX, schrittwesie Reduktion der Kortikosteroide, alternativ Rituximab
Mikroskopische Polyangiitis (MPA)
- wie bei Wegenerscher Granulomatose
Churg-Strauss-Syndrom (eosinophile Granulomatose mit Polyangiitis –EPGA)
- Therapie des Asthma bronchiale
- bei Manifestationen an Herz, Niere, ZNS Therapie wie bei Wegenerscher Granulomatose
- -IL-5-Antagonsit Mepolizumab in klinischer Erprobung
IgA-Vaskulitis (Purpura Schönlein-Henoch – PSH)
- Glukokortikoide
- bei Proteinurie zusätzlich ACE-Hemmer/AT1-Antagonisten
- bei schwerem Verlauf zusätzlich Cyclophosphamid
Kryoglobulinämische Vaskulitis
- Behandlung der Grundkrankheit im Vordergrund (bei HCV-Infektion antivirale Therapie)
- MTX
- bei progredientem Verlauf Cyclophosphamid + Kortikosteroide
- Rituximab als Reservemittel
Hypokomplementämische urtikarielle Vaskulitis
- Prednisolon
- Immunsuppressiva je nach Schwere des Krankheitsbildes
Cogan-Syndrom
- Kortikosteroide, MTX, Azathioprin, ggf. Cyclophosphamid
Morbus Behcet
- Kortikosteroide
- Colchizin bei leichten Fällen
- ggf. Immunsuppressiva (Azathioprin, Ciclosporin), bei lebensbedrohlicher Manifestation auch Cyclophosphamid
- bei Augenbeteiligung IFNα2a
- Reservemedikament bei therapieresistenten Formen: TNFα-Blocker
- Experimentell bei Aphthen: Apremilast
Bei Stenosen können ggf. Revaskularisierungsmaßnahmen erforderlich sein.
Eine wirksame Prävention ist nicht bekannt.
1. Herold - Innere Medizin 2017
Leitlinie der Gesellschaft für Kinder- und Jugendrheumatologie und der Deutschen Gesellschaft für Kinder- und Jugendmedizin:
Weitere Vaskulitiden (Stand 01/2013)
Leitlinie der Deutschen Gesellschaft für Neurologie:
Zerebrale Vaskulitis und zerebrale Beteiligung bei systemischen Vaskulitiden und rheumatischen Grunderkrankungen (Stand 04/2018)


Verschenken Sie kein Honorar: Das „Gebühren-Handbuch digital“ ist die ideale Weiterentwicklung der Printausgabe des bekannten „Medical Tribune Gebühren-Handbuchs“ - statt 2000 Buchseiten der schnelle digitale Zugriff.
Was Ihnen die Abrechnung leichter macht:
- die immer aktuelle Fassung von EBM und GOÄ (Einheitlicher Bewertungsmaßstab und Gebührenordnung für Ärzte)
- Tipps und Experten-Kommentare zur Honorarabrechnung (EBM/GOÄ), graphisch aufbereitet und leicht verständlich
- Kommentare von Kollegen lesen und selbst kommentieren
- persönliche Notizen und Lesezeichen setzen
Fortbildungen
| Termin | Fortbildung | Ort | |
|---|---|---|---|
|
08.11.2025 | 10:00 - 17:00
|
BPC-Fachsymposium Cannabinoide in der Medizin 2025 4. Fachsymposium: Cannabinoide in der Medizin Details Präsenz-Teilnahme Online-Teilnahme |
Düsseldorf |
CME Punkte werden beantragt
kostenfrei
|
Diese Informationen dienen ausschließlich der Aus- und Weiterbildung von Angehörigen und Studenten der medizinischen Fachkreise (z.B. Ärzte) und enthalten nur allgemeine Hinweise. Sie dürfen nicht zur Selbstdiagnose oder -behandlung verwendet werden und sind kein Ersatz für eine ärztliche Beratung oder Behandlung. Die jeweiligen Autoren haben die Inhalte nach bestem Wissen gepflegt. Dennoch sollten Sie die Informationen stets kritisch prüfen und mit zusätzlichen Quellen vergleichen. Die Autoren und die Betreiber von medical-tribune.de übernehmen keine Haftung für Schäden, die durch nicht-kontrollierte Anwendung von Empfehlungen und Inhalten entstehen. Beiträge, die Angaben zum Einsatz und zur Dosierung von Medikamenten machen, sind die persönliche Einschätzung der Autoren. Sie ersetzen nicht die Empfehlungen des Herstellers oder des behandelnden Arztes oder Apothekers.