Kupferspeicherkrankheit: Ablagerungen in der Hornhaut deuten auf Morbus Wilson
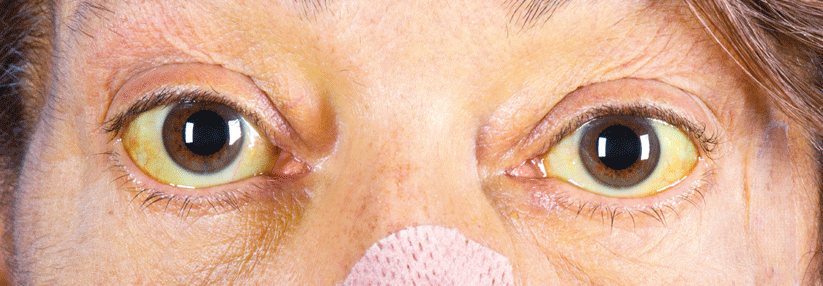
 Typisch für die Krankheit sind Kupferablagerungen in der Hornhaut, die als sogenannter Kayser-Fleischer-Kornealring sichtbar werden.
© iStock.com/lvcandy und wikimedia/Herbert L. Fred, MD, Hendrik A. van Dijk (CC BY 3.0)
Typisch für die Krankheit sind Kupferablagerungen in der Hornhaut, die als sogenannter Kayser-Fleischer-Kornealring sichtbar werden.
© iStock.com/lvcandy und wikimedia/Herbert L. Fred, MD, Hendrik A. van Dijk (CC BY 3.0)
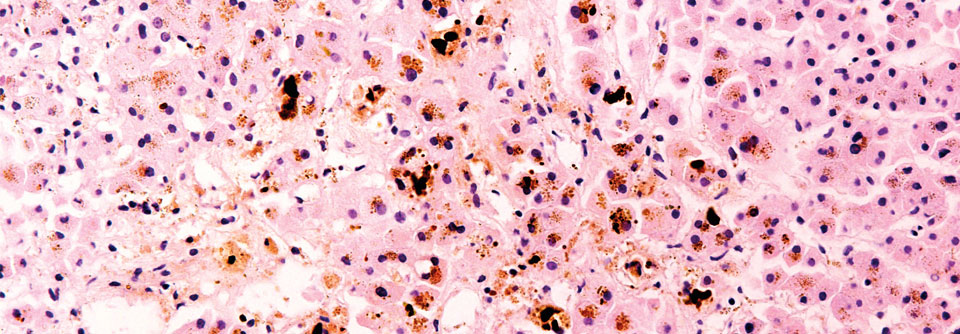
Morbus Wilson ist eine Stoffwechselerkrankung, bei der es aufgrund einer gestörten Kupferausscheidung zu einer Kupferakkumulation in den Leberzellen und anderen Organen kommt. Dem Ganzen zugrunde liegt eine Mutation im ATP7B-Gen. Wegen der diversen klinischen Erscheinungsbilder und den unterschiedlichen Verläufen gilt die Erkrankung als Chamäleon der Hepatologie – und wird im Praxisalltag häufig übersehen. Etwa die Hälfte der Patienten leidet zum Zeitpunkt der Diagnose bereits an einer Zirrhose, 5 % gar unter akutem Leberversagen.
Die Symptome beschränken sich keineswegs auf Leber und Gehirn, so Bernd Mensing von der Psychiatrischen Universitätsklinik Zürich und seine Kollegen. Daneben kann es zu verschiedenen psychiatrischen Anzeichen wie Störungen in Kognition sowie neurologischen Beschwerden kommen, z.B. Tremor, Rigor und Dystonien. Ebenso trifft man auf ophthalmologische Manifestationen (s. Kasten). Typisch zudem: die coombs-negative hämolytische Anämie, insbesondere bei akutem Leberversagen.
Diagnose auf den zweiten Blick
Zur umfassenden Diagnostik bei Morbus Wilson dürfen die Augen nicht vergessen werden. Typisch für die Krankheit sind Kupferablagerungen in der Hornhaut, die als sogenannter Kayser-Fleischer-Kornealring sichtbar werden. Das Problem dabei: in seltenen Fällen zeigt sich dieser auch im Rahmen anderer cholestatischer Leberleiden, reicht also nicht als alleiniges Diagnosekriterium der Wilson-Krankheit. Leberwerte, Lebersynthese- und Cholestaseparameter, ein Differenzialblutbild, LDH, Haptoglobin, Coeruloplasmin und der Kupfergehalt im Serum sowie im 24-h-Urin vervollständigen das Bild. Werte von > 100 µg Kupfer/24 h (entsprechend > 1,6 µmol Kupfer/24 h) ergeben die Diagnose Morbus Wilson. Als invasiver diagnostischer Test ist gegebenenfalls eine Leberpunktion sinnvoll.
Etwa eine von 90 Personen ist Träger des Gendefekts, das autosomal-rezessiv vererbt wird. Die Wilson-Krankheit manifestiert sich meist vor dem 35. Lebensjahr, kann allerdings auch bei älteren Patienten auftreten. Bei Kindern können unerklärte Konzentrationsstörungen bzw. beeinträchtigte Schulleistungen auf Morbus Wilson hinweisen, schreiben die Autoren.
Diagnostik
Einen spezifischen Test für M. Wilson gibt es nicht. Entsprechend knifflig und komplex gestaltet sich die Diagnostik. Neben hepatologischen, neurologischen und psychiatrischen Zeichen kommt der Familienanamnese eine große Bedeutung zu. Bei der klinischen Untersuchung liegt der Fokus auf Symptomen der chronischen Hepatopathie und der Leberzirrhose (Ikterus, Spider Nävi, Ödeme). Erhärtet sich der Verdacht, sollte man Fachärzte für Hepatologie und Neurologie hinzuziehen, betonen Bernd Mensing und Kollegen. Sie raten außerdem dazu, bei neurologischen Auffälligkeiten eine Magnetresonanztomographie des Gehirns durchzuführen. Damit lassen sich oft Strukturdefekte in den Basalganglien finden. In klinisch unklaren Fällen kann man den Leipzig-Score zur Diagnostik hinzuziehen. Spätestens dann sollte man das familiäre Umfeld des Betroffenen auf eine Erkrankung hin untersuchen.Therapie
Medikamentös wird versucht, die aus dem Gleichgewicht geratene Kupfer-Homöostase wiederherzustellen. Überschüssiges freies Kupfer im Serum wird mittels Chelatoren gebunden, sodass dieses mit dem Urin ausgeschieden werden kann (Induktionsphase). In der anschließenden Erhaltungsphase drosseln Chelatoren oder Zinksalze die Zufuhr neuen Kupfers durch Komplexbildung im Darm. Dabei ist zu beachten, dass die Therapie mit Chelatoren vor allem bei Patienten mit neurologischen Manifestationen zu irreversiblen Schäden oder einer Verschlechterung der Symptome führen kann. Zinksalze gelten in diesen Fällen als die bessere Alternative. Die pharmakologische Behandlung erfolgt lebenslang. Da eine Unterbrechung schwere, mitunter tödliche Komplikationen nach sich ziehen kann, ist eine gute Adhärenz das A und O. Asymptomatische Mutationsträger erhalten eine prophylaktische Therapie in reduzierter Dosierung. Zusätzlich lohnt es, Patienten auf eine kupferarme Diät umzustellen.Monitoring
Regelmäßige Kontrollen sind wesentlich. Die Kupferausscheidung im Urin (nach zweitägiger Therapiepause) gibt Auskunft über den Therapieerfolg, unter Therapie mit Zink lässt sich die Adhärenz durch Messung der Zinkausscheidung kontrollieren, schreiben die Autoren. Patienten mit Leberzirrhose und/oder neurologischen Symptomen sollte man zunächst alle ein bis zwei Wochen einbestellen. Hat sich der Verlauf stabilisiert, reicht es, im ersten Therapiejahr alle drei Monate, im zweiten alle sechs Monate zu kontrollieren. Leberwerte und Leberfunktionsparameter normalisieren sich unter Therapie meist binnen 6–18 Monaten. Muss die Leber aufgrund eines akuten Leberversagens transplantiert werden, führt dies zu einer Heilung des zugrunde liegenden Gendefekts in der Leber. Bei Patienten mit neurologischer Symptomatik dauert es häufig sehr lange, bis sich die Beschwerden bessern.Quelle: Mensing B et al. Ther Umsch 2018; 75: 241-248