Akute hepatische Porphyrien Steckt eine Porphyrie dahinter?
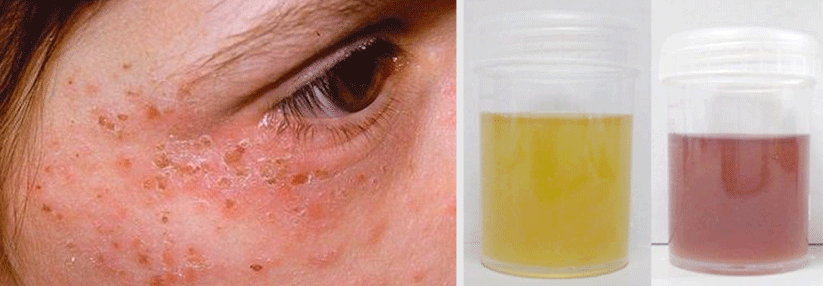
 Für die initiale Diagnostik reicht die Analyse des Spontanurins. Daran schließt sich die Genanalyse an.
© Henrik Dolle – stock.adobe.com
Für die initiale Diagnostik reicht die Analyse des Spontanurins. Daran schließt sich die Genanalyse an.
© Henrik Dolle – stock.adobe.com
Die Gemeinsamkeit der akuten hepatischen Porphyrien (AHP) besteht in der hereditär bedingten Störung der Hämsynthese. Dabei kommt zu einer pathologischen Akkumulation der Präkursoren Deltaaminolävulinsäure (ALA) und Porphobilinogen (PGB).
Unterschieden werden vier Manifestationsformen, am häufigsten ist die akute intermittierende Porphyrie (AIP). Zu den wichtigsten Symptomen von AHP zählen akut einsetzende schwere Bauchschmerzen, Nausea, Erbrechen, Muskelschwäche, Neuropathie, Tachykardie und Hypertonie. Deshalb sollte man bei allen Patienten mit rezidivierenden, aber unerklärlichen abdominellen Schmerzattacken auch an eine akute hepatische Porphyrie denken. Besonders häufig betroffen sind Frauen im Alter zwischen 15 und 50 Jahren, schreiben Dr. Bruce Wang von der University of California in San Francisco und Kollegen.
Vier Typen der AHP
-
Akute intermittierende Porphyrie
-
Hereditäre Koproporphyrie
-
Porphoria variegata
-
Porphyrie mit schwerem Mangel an 5-Aminolävulinsäure
Für die initiale Diagnostik akuter hepatischer Porphyrien wird eine Kontrolle von Deltaaminolävulinsäure (ALA) und Porphobilinogen (PBG) im Spontanurin empfohlen, beide Werte sind während der akuten Attacken um mindestens das Fünffache des oberen Normwerts erhöht, sie können aber auch Monate oder Jahre danach noch gesteigert sein. Zur Korrektur auf die Harnkonzentration ist zusätzlich der Kreatininwert zu erfassen. Die Messung der Gesamtporphyrine im Harn eignet sich nicht zum Screening. Bei einer singulären Erhöhung der ALA sind Bleivergiftung und hereditäre Tyrosinämie auszuschließen.
Wenn die biochemischen Tests positiv ausfallen, empfehlen die Autoren eine genetische Sicherung der Diagnose. Damit lassen sich fast alle AHP-Fälle identifizieren. Erstgradige Verwandte von Betroffenen sollten ebenfalls untersucht werden, sobald die auslösende Genvariante identifiziert ist. So kann man Personen mit erhöhtem Risiko für Porphyrieattacken aufspüren. Die Genanalyse eignet sich aber nicht zum initialen Screening auf eine AHP, denn die meisten Mutationsträger erleiden zeitlebens keine Anfälle.
Infusion einmal täglich über insgesamt vier Tage
Das Management der akuten hepatischen Porphyrie hängt vom Schweregrad ab. Patienten mit stationär behandlungspflichtigen Attacken sollten eine Hämin-Therapie erhalten, die die Überproduktion und Akkumulation von ALA und PBG verringert. Die Infusionen werden einmal täglich verabreicht, üblicherweise über einen Zeitraum von vier Tagen. Wegen des Risikos für eine häminbedingte Thrombophlebitis wird die Applikation über einen zentralen Venenkatheter oder Port empfohlen.
Einen hohen Stellenwert hat die symptomatische Therapie der akuten Anfälle. An erster Stelle steht auch hier die Reduktion der Aminolävulinsäurebildung. Deshalb sollten über Cytochrom P450 verstoffwechselte Arzneimittel sofern möglich abgesetzt werden. Schmerz und Übelkeit lassen sich mit einer intensiven medikamentösen Behandlung lindern. Im Frühstadium der Anfälle kann eine intravenöse Kohlenhydratgabe hilfreich sein. Auch Hypertension, Tachykardie und Hyponatriämie müssen, sofern vorhanden, korrigiert werden. Außerdem sind Triggerfaktoren wie Stress, übermäßiger Alkoholgenuss und Mangelernährung zu vermeiden.
Bei Porphyriepatienten mit mindestens vier Anfällen im Jahr plädieren die Autoren für eine Prophylaxe. Betroffen sind häufig Frauen mit zyklusabhängigen Beschwerden, sie können von einer Hormonsuppression (z.B. GnRH-Agonisten) profitieren. Ansonsten werden vielfach die in der Akuttherapie der AHP bewährten Hämin-Infusionen genutzt. Deren prophylaktische Wirkung ist aber weniger gut belegt. Außerdem muss man mit Komplikationen wie Infektion und Eisenüberlastung rechnen.
Neuartige RNA-basierte Therapieoption zugelassen
Aber es gibt eine neue Therapieoption: Kürzlich wurde in Europa Givorisan zur Prophylaxe bei akuten hepatischen Porphyrien zugelassen. Dabei handelt es sich um eine neuartige RNA-basierte Behandlung, die die Produktion der Aminolävulinsäure reduziert. In einer doppelblinden Phase-3-Studie verminderte das einmal monatlich subkutan gespritzte Givorisan die Zahl der Attacken, ein erheblicher Teil der Patienten wurde sogar anfallsfrei.
Als Ultima Ratio gilt nach wie vor die Lebertransplantation. Sie kommt für Patienten mit therapierefraktären Beschwerden und stark verminderter Lebensqualität in Betracht.
Wichtig ist auch die frühzeitige Detektion porphyriebedingter Organschäden. So entwickeln Patienten mit AHP häufiger eine Leberfibrose und Zirrhose. Deshalb wird eine jährliche Kontrolle empfohlen. Auch eine dauerhafte Hämin-Therapie kann via Eisenüberladung eine Fibrose auslösen. Deswegen sollten die Leberenzyme mindestens jährlich bestimmt werden, Ferritin und Eisenwerte alle drei bis sechs Monate.
Außerdem besteht ein erhöhtes Risiko für hepatozelluläre Karzinome (HCC). Diese treten bei Frauen mehr als doppelt so häufig auf wie bei Männern. Sie entwickeln sich auch ohne vorbestehende Fibrose und sogar ohne Porphyriesymptome. Die Autoren plädieren deshalb dafür, auch AHP-Patienten ohne Beschwerden ab einem Alter von 50 Jahren alle sechs Monate sonografisch zu untersuchen.
Das Risiko für eine chronische Nierenerkrankung ist bei der AHP ebenfalls gesteigert, deswegen wird ein jährliches Screening (Kreatinin, eGFR) angeraten. Eine weitere häufige Folge der Porphyrie ist die arterielle Hypertonie. Sie tritt zunächst während der Attacken auf, kann aber chronifizieren. Umso wichtiger sind regelmäßige Blutdruckkontrollen.
Quelle: Wang B et al. J Gastroenterol 2023; 164: 484-491; DOI: 10.1053/j.gastro.2022.11.034