Ursachen der Lungenfibrose Schritt für Schritt ausschließen – bis „idiopathisch“ übrig bleibt
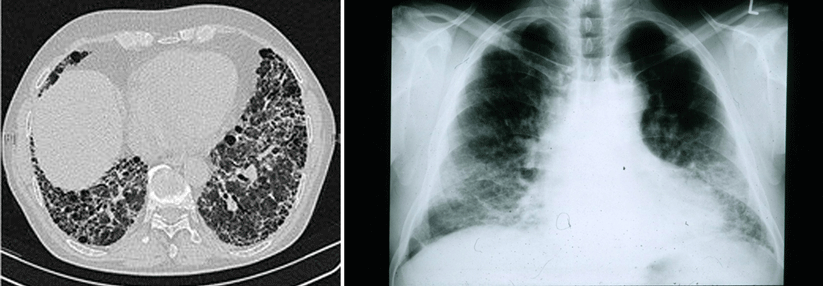
 Die Bildgebung mit HRCT lässt sich anhand von vier Diagnosekategorien beurteilen.
© iStock/invincible_bulldog
Die Bildgebung mit HRCT lässt sich anhand von vier Diagnosekategorien beurteilen.
© iStock/invincible_bulldog
Eine hohe diagnostische Sicherheit bei möglichst geringer Invasivität der Untersuchungsmethoden – so lautet der Tenor der aktuellen S2K-Leitlinie zur idiopathischen pulmonalen Fibrose (IPF). Und natürlich strebt man eine frühe Entdeckung der Erkrankung an.
Als bekannte Ursachen für den chronisch progredienten Umbau des Lungenparenchyms kommen u.a. folgende Erkrankungen infrage:
- Autoimmunerkrankungen mit Lungenbeteiligung, vor allem mit rheumatoider Arthritis oder anderen Kollagenosen assoziierte interstitielle Lungenerkrankungen (ILD)
- medikamentös bedingte Pneumopathien
- Pneumokoniosen
- chronische exogen allergische Alveolitis (EAA)
- chronische Sarkoidose
- selten Infektionen (z.B. Tbc)
Die Leitlinienautoren unter Koordination von Professor Dr. Jürgen Behr von der Medizinischen Klinik V an den Asklepios Fachkliniken Gauting haben nun wichtige Grundprinzipen der IPF-Diagnostik und Differenzialdiagnostik überprüft und Empfehlungen formuliert.
Der Weg zur IPF-Diagnose
- Ausschluss einer ILD bekannter Ursache (z.B. durch Exposition gegenüber inhalativen Noxen, Kollagenosen und andere Systemerkrankungen, medikamenteninduzierte ILD etc.) plus entweder 2 oder 3
- Vorhandensein eines UIP-Musters im HRCT
- Spezifische Kombinationen von HRCT und Histologie
Wonach ist in der Anamnese zu fragen?
Wichtig ist z.B. die Frage nach dem Rauchen. 60-70 % der Betroffenen werden sie bejahen. Die IPF manifestiert sich allerdings fast immer Jahre bis Jahrzehnte nach dem Rauchstopp. Auch die inhalative Exposition gegenüber Asbest, Metall- und Holzstäuben, Chemikalien oder Allergenen sowie die Medikation der Patienten müssen erfasst werden.Was bringen serologische Tests?
Sie sollten bei Verdacht auf eine IPF regelhaft durchgeführt werden, um Kollagenosen als mögliche Differenzialdiagnosen zu identifizieren. Spezielle Biomarker, die der Abgrenzung der IPF von anderen ILD dienen sollen, haben sich bisher nicht als hilfreich erwiesen, die Leitlinie rät von der Messung daher ab.Welche Bildgebung hat die größte Bedeutung?
Die Experten nennen an erster Stelle die hochauflösende Computertomographie (HRCT), die in jedem IPF-Verdachtsfall zum Einsatz kommen sollte. Empfohlen wird die Untersuchung ohne Röntgenkontrastmittel in Inspiration und Rückenlage. Will man eine Erkrankung der kleinen Atemwege wie eine Bronchiolitis oder eine EAA abklären, helfen zusätzlich sequenzielle HRCT-Schichtaufnahmen in Exspiration. Je nach HRCT-Befund ergeben sich vier Diagnosekategorien:- UIP-Muster: UIP = gewöhnliche interstitielle Pneumonie; charakteristisch für die IPF, Honigwaben mit oder ohne periphere Traktionsbronchiektasien
- wahrscheinliches UIP-Muster: subpleurale, basal-betonte retikuläre Veränderungen mit peripherer Traktionsbronchiektase oder Bronchiolektase
- unbestimmt für UIP-Muster: Etwa jeder dritte Patient mit histopathologisch gesichertem UIP-Muster bietet im HRCT atypische Befunde, aber meist Zeichen einer Fibrose. Unter die Kategorie „unbestimmt“ fallen auch Patienten mit Verdacht auf eine frühe UIP oder wahrscheinliche UIP (scharf begrenzte subpleurale Milchglastrübung oder Retikulation, keine klaren Kennzeichen einer Fibrose).
- alternatives Muster
Wie geht es nach der Bildgebung weiter?
Abhängig von den verschiedenen anamnestischen, klinischen Befunden und dem Ergebnis der HRCT geben die Leitlinien-Experten folgende Empfehlungen für die weitere Abklärung von Patienten mit Verdacht auf eine idiopathische Lungenfibrose:- UIP-Muster in der HRCT, keine klinischen Hinweise auf eine andere ILD: keine bronchoalveoläre Lavage (BAL), keine Biopsie.
- UIP-Muster in der HRCT mit klinischen Hinweisen auf eine andere ILD: BAL
- wahrscheinliches UIP-Muster in der HRCT, Alter über 60 Jahre, männliches Geschlecht, Exraucher: BAL, aber keine Biopsie. Ohne die drei letzten Kriterien: BAL plus Kryobiopsie
- unbestimmt für UIP oder alternatives HRCT-Muster: BAL und Kryobiopsie
- Bei weiterhin unklarer Diagnose nach Endoskopie und multidiziplinärer Diagnostik: chirurgische Lungenbiopsie
Chirurgisch oder transbronchial biopsieren?
Im Unterschied zu internationalen Leitlinien gibt die deutsche der Kryobiopsie gegenüber der chirurgischen Biopsie den Vorrang. Sie habe keine Nachteile hinsichtlich der Aussagekraft, sei kostengünstiger und stoße auf mehr Patientenakzeptanz, heißt es in der Publikation. Zudem könnten auch Menschen mit fortgeschrittener interstitieller Lungenerkrankung, mehreren Komorbiditäten und in höherem Alter kryobiopsiert werden.
Die Autoren hoffen, dass sich durch den einfacheren Einsatz dieses Verfahrens der Anteil unklassifizierbarer ILD verringert. Sie geben allerdings auch zu, dass die Methode bisher nicht international standardisiert ist und der Vergleich der beiden Biopsiemethoden auf schwacher Evidenz beruht.
Die Histopathologie orientiert sich ebenfalls an den vier oben genannten HRCT-Diagnosekategorien.
Anhand der invasiven Diagnostik sollte gemäß der Leitlinie dann nochmals multidisziplinär die endgültige Diagnose und das weitere Vorgehen festgelegt werden.
Quelle: S2K-Leitlinie zur Diagnostik der Idiopathischen Lungenfibrose. AWMF-Register Nr. 020/016; www.awmf.org