Idiopathische Lungenfibrose: Wie soll die Frühdiagnostik gelingen?
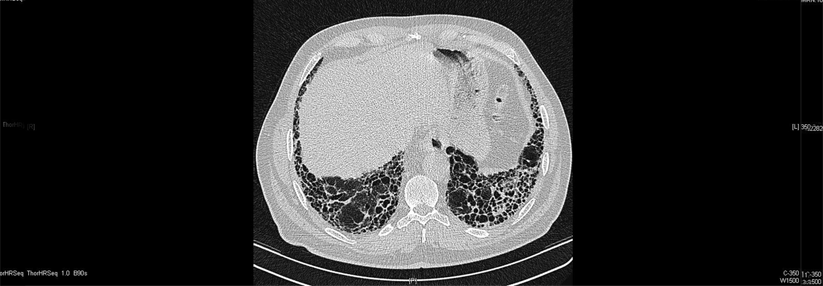
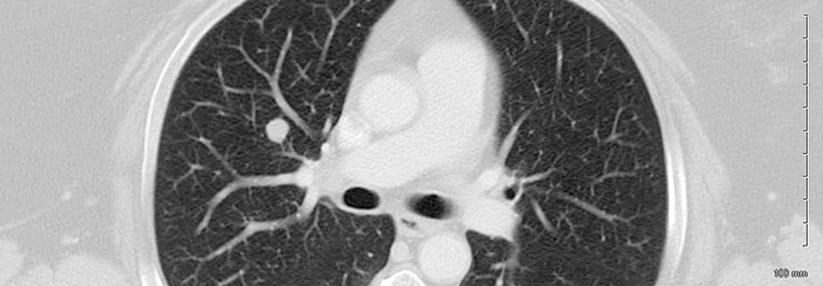
 Nicht immer lässt sich eine IPF wie hier erkennen. Oft scheitert das hochauflösende CT an Veränderungen < 2 mm und in frühen Stadien.
© wikimedia/IPFeditor (CC BY-SA 3.0)
Nicht immer lässt sich eine IPF wie hier erkennen. Oft scheitert das hochauflösende CT an Veränderungen < 2 mm und in frühen Stadien.
© wikimedia/IPFeditor (CC BY-SA 3.0)
Auffällige interstitielle Lungenbefunde (ILA) im CT sind vor allem bei älteren Menschen ein häufiger Zufallsbefund, der mit Einführung des Lungenkrebsscreenings noch öfter anfallen dürfte. Die Bewertung im Einzelfall ist jedoch schwierig, ob tatsächlich eine fortschreitende Fibrose mit raschem pulmonalem Funktionsverlust droht. Professor Dr. Gary M. Hunninghake, Harvard University, Boston, forderte, zu standardisieren, wie solche radiologischen Befunde erhoben und berichtet werden – das würde als Nebeneffekt auch Forschungsergebnisse einfacher vergleichbar machen. Außerdem müsse ein Konsensus entwickelt werden, wann solche Normabweichungen auch bei asymptomatischen Patienten als Krankheit gewertet werden sollten. Ein erstes Positionspapier, wie Zufallsbefunde zu beurteilen sind, hat die Fleischner Society gerade veröffentlicht.1
Noch kein Verdacht auf ILD
Als ILA werden darin retikuläre Zeichnungen, fleckige Infiltrate oder Knötchen, Honeycombing, Milchglastrübungen, Traktionsbronchiektasen und nicht-emphysematöse Zysten definiert, die mindestens 5 % eines Lungenabschnitts einnehmen. Wichtige Neuerung im Vergleich zu früheren Definitionen: Sie beschränkt sich explizit auf Patienten, bei denen bislang kein Verdacht auf eine interstitielle Lungenerkrankung (ILD) besteht. Ferner soll danach differenziert werden, ob sich die ILA vorwiegend subpleural befinden oder über die Lunge verteilen und ob fibrotische Veränderungen vorliegen oder „nur“ retikuläre oder Milchglasveränderungen.
Nachdem im vergangenen Jahr eine Studie gezeigt hatte, dass ILA-Patienten mit wahrscheinlichen oder definitiven UIP-Pattern ein erhöhtes Progressionsrisiko haben, sollten diese einem „aktiven Monitoring“ zugeführt werden. Das bedeutet:
- Risikofaktoren identifizieren und behandeln,
- alle drei bis zwölf Monate zu klinischer Untersuchung und Lungenfunktionstest einbestellen,
- CT alle 12–24 Monate wiederholen.
Die Zeiträume können auch kürzer ausfallen, wenn Klinik oder Befinden einen Progress vermuten lassen. Bei Patienten ohne UIP-Muster kann man sich auf Empfehlungen zu Risikofaktoren beschränken. Nachuntersuchungen sind erst fällig, wenn Symptome auftreten.
Ohne diagnostischen Overkill Betroffene identifizieren
Eine Herausforderung liegt darin, ein Screening für Hochrisikopatienten zu entwickeln, das Betroffene ohne diagnostischen Overkill identifiziert. Möglicherweise kann die immer breiter verfügbare Gendiagnostik dabei helfen, meinte Prof. Hunninghake. Es gibt bereits Genpanel, die typisch sind für Patienten mit interstitieller Lungenfibrose (IPF), die sich teilweise mit denen von ILA-Patienten überschneiden, aber auch einige wichtige Unterschiede zeigen.
Prof. Hunninghakes Team hat ein Pilotprogramm bei erstgradig Verwandten von Lungenfibrose-Patienten gestartet, zu dem neben klinischer Untersuchung, Labor, CT und Immunphänotypisierung auch das Messen der Telomerlänge sowie gezielte Genotypisierung und Gensequenzierung gehören.2 Von 105 Teilnehmern aus 53 Familien hatten 31 % ILA und 18 % eine manifeste interstitielle Lungenerkrankung (ILD). Dabei machte es keinen Unterschied, ob die Lungenfibrose in der Familie lag oder sporadisch aufgetreten war.
Neue Faktoren für die Risikoabschätzung
Die Forscher konnten Faktoren identifizieren, die mit einer erhöhten ILA/ILD-Wahrscheinlichkeit assoziiert waren: schlechte Lungenfunktion (Vital-, Gesamtlungen- oder Diffusionskapazität) sowie auf Genebene reduzierte Telomerlänge der Lymphozyten und mehr Kopien der MUC5B-Promotervariante. Sie verbesserten die Risikoabschätzung erheblich, wenn man sie zu Alter, Geschlecht, Familien- und Rauchanamnese hinzunahm.
Eine neue Strategie für die frühe Diagnose der IPF als prognostisch ungünstigste Variante der interstitiellen Lungenerkankungen stellte Professor Dr. Lida P. Hariri, Massachusetts General Hospital, Boston, vor. Bislang ist das hochauflösende CT der Goldstandard, scheitert aber oft an sehr kleinen Veränderungen unter 2 mm und in frühen Stadien, sodass eine Lungenbiopsie mit all ihren Komplikationsrisiken in Kauf genommen werden muss.
Die optische Kohärenztomographie (OCT) könnte eine Alternative sein. Sie funktioniert ähnlich wie Ultraschall, arbeitet aber mit Licht- statt Schallwellen. Sie wird z.B. routinemäßig eingesetzt, um mikroskopisch kleine Veränderungen am Augenhintergrund sichtbar zu machen.
Eine Anwendung per Bronchoskop liefert blitzschnell Bilder in Echtzeit, sodass sich große Teile der Lunge bis hin zu subpleuralen Bereichen in kurzer Zeit untersuchen lassen.
Prof. Hariris Arbeitsgruppe hat bereits diagnostische Kriterien erarbeitet, mit denen sich UIP- und NSIP-Muster von gesunden Lungen und untereinander abgrenzen lassen. Prospektive Vergleiche mit den Ergebnissen der späteren Lungenbiopsie von 27 ILD-verdächtigen Patienten ergaben ein hohes Maß an Übereinstimmung.3 Alle, bei denen die OCT ein UIP-Muster gezeigt hatte, erhielten später die Diagnose IPF. Bei allen, bei denen die OCT kein UIP-Muster gefunden hatte, wurde histologisch eine andere ILD diagnostiziert. Daraus ergibt sich eine Sensitivität und Spezifizität für UIP von 100 %. Die OCT könnte demnach eine frühere Diagnose liefern, zum Beispiel nach dem initialen hrCT – ganz ohne Operation und weitere Strahlenexposition. Außerdem bietet sie natürlich die Möglichkeit einfacher nicht-invasiver Verlaufskontrollen.
Mehr Forschung durch Früherkennung
Auch hinsichtlich früher Interventionen bleiben viele Fragen zu klären, räumte Prof. Hunninghake ein. Welche Optionen stehen überhaupt zur Verfügung, pharmakologisch wie nicht-pharmakologisch, und was bewirken sie in frühen Krankheitsstadien? Gibt es Prozesse, die auf das frühe Stadium beschränkt sind und sich gezielt – mit noch zu entwickelnden Strategien – angehen lassen? Eine bessere Frühdiagnostik böte die Chance, diesen Fragen nachzugehen.
* American Thoracic Society
Quellen:
1. Hatabu H et al. Lancet Respir Med 2020; 8: 726-737; DOI: 10.1016/S2213-2600(20)30168-5
2. Hunninghake GM. AJRCCM 2020; 201: 1240-1248; DOI: 10.1164/rccm.201908-1571OC
3. Hariri LP et al. AJRCCM 2018; 197: 949-952; DOI: 10.1164/rccm.201707-1446LE.