Lynch-Syndrom verursacht 3 % aller kolorektalen Karzinome
 50 % der Nachfahren tragen dieselbe Mutation.
© iStock.com/Mohammed Haneefa Nizamudeen
50 % der Nachfahren tragen dieselbe Mutation.
© iStock.com/Mohammed Haneefa Nizamudeen
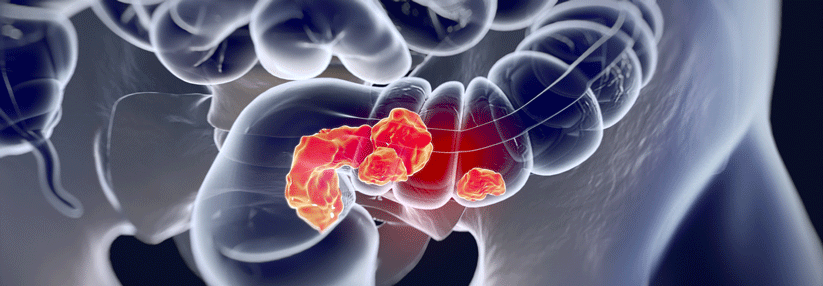
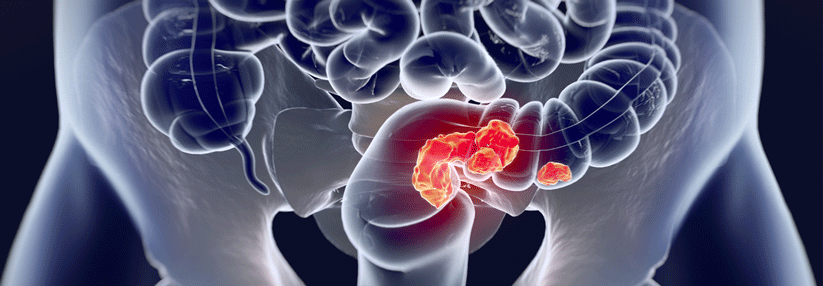
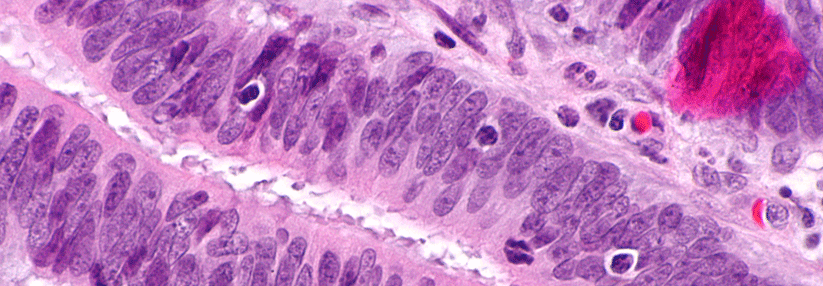
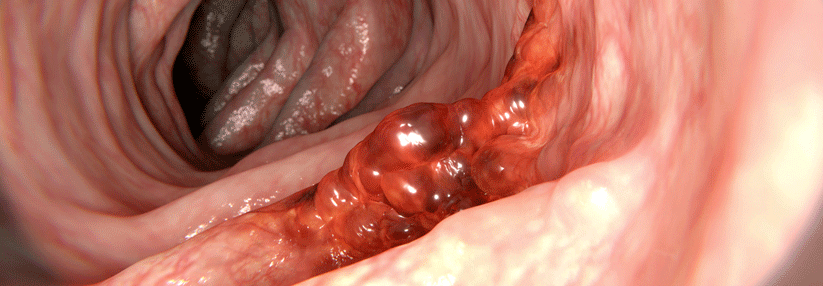
Gekennzeichnet ist der Lynch-Syndrom-Phänotyp durch einen überwiegend auf der rechten Seite des Dickdarms vorkommenden Krebs sowie eine Neigung zu synchronen und metachronen kolorektalen Karzinomen. Es macht 3 % aller Neudiagnosen des kolorektalen Karzinoms aus. Die Tumoren zeigen typischerweise eine schlechte Differenzierung, die muzinöse Merkmale oder ein medulläres Wachstumsmuster umfassen kann. Obwohl Dickdarmkrebs die häufigste Krebsart ist, die mit dem Lynch-Syndrom in Verbindung gebracht wird, können auch extrakolonale Krebsarten auftreten, darunter Endometriumkarzinome, Tumoren im Dünndarm, Ureter, Nierenbecken, Magen, hepatobiliären Trakt und Eierstock.
Diagnose beeinflusst das Ausmaß der Kolonresektion
Die mangelhafte Reparatur von Fehlpaarungen führt zur Hypermutation. Kolonkarzinome mit defekten Reparaturgenen sind mit einem frühen Erkrankungsalter und einer geringen Metastasenneigung assoziiert. Studien zufolge haben Patienten mit nicht-metastasiertem Darmkrebs und fehlerhaftem Mismatch-Repair eine signifikant bessere Prognose als Patienten mit Tumoren desselben Stadiums, aber funktionierender Mismatch-Reparatur, zeigt Dr. Frank A. Sinicrope vom Mayo Comprehensive Cancer Center in Rochester auf.
Die Diagnose des Lynch-Syndroms wird durch die Identifizierung einer Keimbahnmutation in einem Mismatch-Reparatur-Gen (MLH1, MSH2, MSH6 oder PMS2) oder einer Keimbahndeletion in EpCAM (epitheliales Zelladhäsionsmolekül) gestellt. Das Krebsrisiko hängt vom betroffenen Gen ab. Mutationen in MLH1 oder MSH2 sind wahrscheinlich für 60–80 % aller Lynch-Syndrom-assoziierten Krebsarten verantwortlich.
Das Lynch-Syndrom unterliegt einem autosomal-dominanten Erbgang. Entsprechend geben betroffene Personen – unabhängig davon, ob sich der Krebs bei ihnen entwickelt – die Mutation zu 50 % an ihre Nachkommen weiter. Wenn in der Familienanamnese eine Person die Amsterdam-I- oder -II-Kriterien erfüllt, ist die Wahrscheinlichkeit einer klinischen Diagnose hoch. Da dies allerdings nur bei der Hälfte aller Betroffenen der Fall ist, wurde mit den revidierten Bethesda-Kriterien die Diagnostik erleichtert.
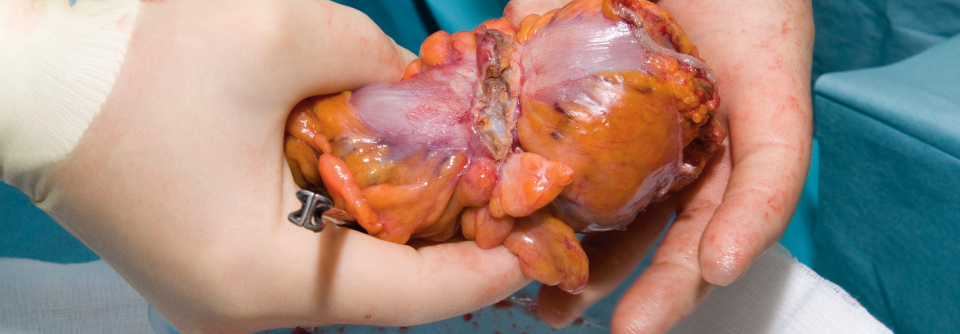
Da die Diagnose das Ausmaß der Kolonresektion beeinflussen kann, sollte vor Einleitung chirurgischer Maßnahmen abgeklärt werden, ob ein Lynch-Syndrom vorliegt.
Klinische Merkmale
- frühes Manifestationsalter
- Lokalisation des Tumors überwiegend im rechten Hemikolon
- häufig extrakolonale Tumormanifestationen
- muzinöse oder siegelringzellige Adenokarzinome mit entzündlicher Infiltration
Teilnahme an Immuntherapie-Studie erwägen
Beobachtungsstudien haben nämlich gezeigt, dass das Risiko eines metachronen kolorektalen Karzinoms bei Patienten mit Lynch-Syndrom, die sich bei der Diagnose eines ersten Kolonkarzinoms einer subtotalen Kolektomie unterzogen hatten, geringer war als bei jenen, bei denen lediglich eine Teilresektion vorgenommen wurde, berichtet der Experte. Patienten mit einem Kolonkarzinom des Stadiums 3 und defekter Fehlpaarungsreparatur können für die Teilnahme an einer klinischen Immuntherapie-Studie in Betracht gezogen werden. Für Patienten mit metastasiertem kolorektalem Karzinom und defekter Fehlpaarungsreparatur sind Immun-Checkpoint-Inhibitoren Standardtherapie.Quelle: Sinicrope FA. N Engl J Med 2018; 379: 764-773