Strategien zur Behandlung des Ewing-Sarkoms
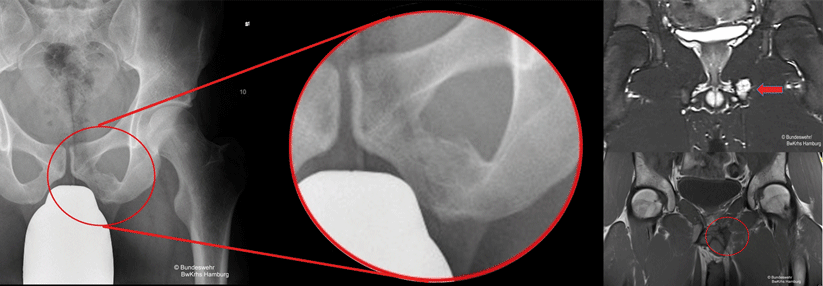
 Speiche mit Ewing-Sarkom. Die Dehnung des Periosts bereitet Schmerzen, die meist belastungsabhängig zunehmen. Diese werden nicht selten als Folge einer Sportverletzung fehlinterpretiert.
© Science Photo Library/Devlin, Mike
Speiche mit Ewing-Sarkom. Die Dehnung des Periosts bereitet Schmerzen, die meist belastungsabhängig zunehmen. Diese werden nicht selten als Folge einer Sportverletzung fehlinterpretiert.
© Science Photo Library/Devlin, Mike
Das Ewing-Sarkom ist der zweithäufigste Knochentumor im Kindesalter, es befällt aber auch junge Erwachsene. Das aggressive Malignom kann sich in jedem Körperteil manifestieren. Besonders oft betroffen sind Becken und lange Röhrenknochen (s. Kasten). Die klinischen Zeichen sind weitgehend unspezifisch, schreiben Dr. Nicolò Riggi von der Universitätsklinik Lausanne und Kollegen. Patienten klagen beispielsweise über leichte Schmerzen, die mit einer Schwellung einhergehen und deshalb fälschlich als Traumafolge gedeutet werden. Manchmal verschlimmern sich die Beschwerden nachts oder nach dem Sport. Auch tastbare Verhärtungen und pathologische Frakturen können das erste Zeichen sein. Im Röntgenbild fallen multiple mottenfraßähnliche Osteolysen und eine zwiebelschalenförmige Abhebung des Periosts auf.
Lokalisation des Ewing-Sarkoms
- Ossärer Primärtumor: Achsenskelett (z.B. Becken, Rippen), distales Skelett (z.B. Femur, Humerus)
- Extraossäre Tumoren: meist paravertebral oder im thorakalen Weichgewebe, seltener in inneren Organen
- Metastasen: vor allem in Lunge, Knochen, Knochenmark
Die Behandlung von Patienten mit primärem Ewing-Sarkom fußt auf einer Kombination von Chemotherapie und lokaler Tumorreduktion (Operation, Radiatio). Dadurch ist das 5-Jahresüberleben von Patienten mit lokal begrenzter Erkrankung von 10 % auf 70 % gestiegen. In Studien wurden verschiedene Formen der Dosis-Intensivierung getestet: Dabei scheint die Verkürzung der Intervalle zwischen den Applikationen wirksamer und weniger toxisch zu sein als die Kombination von Hochdosis-Chemotherapie und Stammzelltransplantation. Allerdings ist das rezidivierte bzw. metastasierte Ewing-Sarkom bis heute nicht kurativ behandelbar. Außerdem drohen nach Radio- und Zytostatikatherapie brisante Sekundärschäden, die vom myelodysplastischen Syndrom bis zum Zweittumor reichen.
Eine an der chromosomalen Genese orientierte Behandlung könnte die Prognose fortgeschrittener Tumoren entscheidend verbessern – ohne die mit den herkömmlichen Methoden verbundene Toxizität. Optimal wäre eine direkte Inhibition des tumorauslösenden Fusionsproteins. Allerdings lässt sich dieser Schritt mit den bisher verfügbaren Techniken nicht durchführen. Also muss man auf alternative Strategien ausweichen. Dazu bietet sich beispielsweise die Hemmung von Effektormolekülen des Fusionsproteins an. Bisher wurden diverse Kandidaten untersucht, darunter auch der Rezeptor für den Insulin-like-Growth-Factor 1 (IGF-IR). Er wird durch EWS-FLI1 induziert und für die Transformation von Fibroblasten benötigt. Gegen IGF-IR gerichtete Antikörper und andere Ansätze zeigten aber nur eine geringe Wirksamkeit.
Relevante Zelltypen gezielt ausschalten
Vielversprechende Ergebnisse lieferten dagegen Untersuchungen auf Einzelzellebene. Sie deuten darauf hin, dass sich bestimmte zelluläre Subpopulationen, die beim Ewing-Sarkom eine Rolle spielen, gezielt ausschalten lassen. Die in Bezug auf das Ewing-Sarkom gewonnenen Erkenntnisse könnten auch neue Möglichkeiten zur Behandlung anderer Tumoren eröffnen, die durch chromosomale Transformationen und aberrierende Fusionsproteine ausgelöst werden, betonen die Kollegen aus der Schweiz.Quelle: Riggi N et al. N Engl J Med 2021; 384: 154-164; DOI: 10.1056/NEJMra2028910
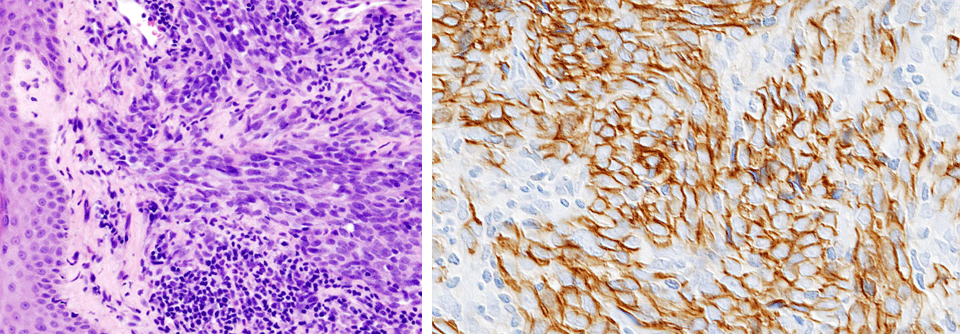
Die definitive Diagnose des Ewing-Sarkoms beruht heute auf dem Nachweis der auslösenden chromosomalen Translokation mittels In-situ-Hybridisierung oder Polymerasekettenreaktion. Am häufigsten ist mit einem Anteil von 85–90 % der Fälle die Translokation t(11;22)(q24;q12). Sie führt zur Produktion des onkogenen Fusionsproteins EWS-FLI1, das in der Pathogenese des Ewing-Sarkoms eine zentrale Rolle spielt.
Der Tumor entwickelt sich wahrscheinlich aus mesenchymalen Stamm- und Stromazellen, die dem Fusionsprotein EWS-FLI1 eine geeignete Umgebung für eine maligne Transformation bieten. Mit der Verschiedenartigkeit dieser Vorläuferzellen für Binde- und Knochengewebe lässt sich auch die auffällige intratumorale Heterogenität des Malignoms erklären.
Wer hat die besseren Aussichten?
Patienten mit Lungenmetastasen haben eine günstigere Prognose als Betroffene mit Absiedelungen in Knochen oder Knochenmark. Beim nicht gestreuten Tumor ist die Lokalisation entscheidend: Im Fall einer primär proximalen Läsion (z.B. Becken, Os sacrum) sind die Aussichten schlechter als bei einer distalen Manifestation. Zu den Risikofaktoren für einen ungünstigen Verlauf gelten zudem ein großer Primärtumor, Patientenalter > 18 Jahre und erhöhte Lakatdehydrogenase-Werte.